Table Of Content
- Computational Design of Novel Enzymes Without Cofactors
- Extended Data Fig. 5 Symmetric oligomer design with RFdiffusion.
- Symmetric Protein Architecture in Protein Design: Top-Down Symmetric Deconstruction
- Computational Redesign of Metalloenzymes for Catalyzing New Reactions
- Directed evolution of a recombinase that excises the provirus of most HIV-1 primary isolates with high specificity
- Constrained peptides
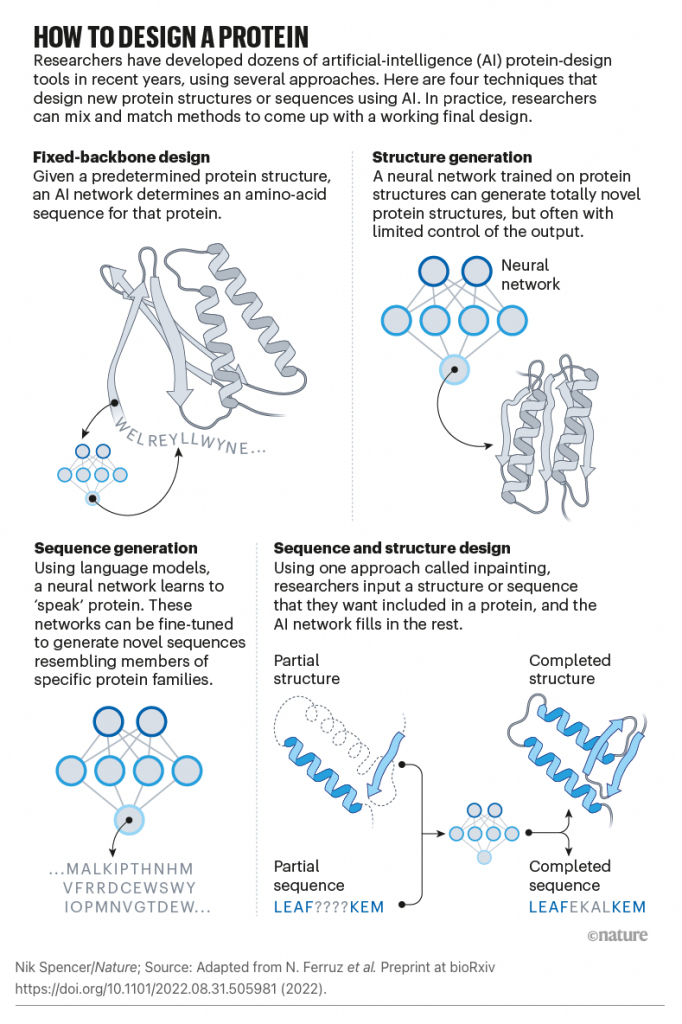
- Hallucinating functional protein sequences
Advanced computational techniques, including novel machine learning algorithms, allow our scientists to model the behavior of proteins at the atomic level. This knowledge helps researchers generate novel proteins with optimized stability, binding affinity, or catalytic activity. Protein design is the process of creating new proteins with specific properties by manipulating the sequence of amino acids. It involves a combination of computational techniques, laboratory experiments, and interdisciplinary knowledge from fields such as biology, chemistry, and physics. To date, companies operating in the protein design space have largely focused on retooling existing proteins to perform new tasks or enhance specific properties, rather than true design from scratch. For example, scientists at Generate Biomedicines have drawn on existing knowledge about the SARS-CoV-2 spike protein and its interactions with the receptor protein ACE2 to design a synthetic protein that can consistently block viral entry across diverse variants.
Computational Design of Novel Enzymes Without Cofactors
We would like to thank the Rosetta community and members of the Kortemme lab for many contributions to computational design and insightful discussions. Whether you’re a student, researcher, partner, or supporter, there’s a place for you in the protein design revolution. Protein TherapeuticsComputer-generated molecules that reprogram cells, block infection, capture toxins, and more. Proteins can do remarkable things, from blocking infection to harnessing solar energy.
New bioengineered protein design shows promise in fighting COVID-19 - Phys.org
New bioengineered protein design shows promise in fighting COVID-19.
Posted: Thu, 14 Mar 2024 07:00:00 GMT [source]
Extended Data Fig. 5 Symmetric oligomer design with RFdiffusion.
Thus, by supplying radioactively labeled ATP derivatives to cells expressing the mutant kinase, only substrates of that particular kinase would be labeled (Figure 17). The key is to engineer stimuli-responsiveness in a regime that is compatible with physiological temperature. Building on the pioneering work of Ho and DeGrado (J Am Chem Soc 1987, 109, 6751–6758) in the late 1980s, protein design approaches have revealed many fundamental features of protein structure and stability. We are now in the era that the early work presaged – the design of new proteins with practical applications and uses. Here we briefly survey some past milestones in protein design, in addition to highlighting recent progress and future aspirations.
Symmetric Protein Architecture in Protein Design: Top-Down Symmetric Deconstruction
Small molecule-based methods are effective, but they are restricted by molecular diffusion through the plasma membrane and cell walls. In principle, light would be an ideal stimulus because illumination can be rapidly switched on or off, resulting in essentially instantaneous addition or removal of signal. Moreover, it can reach any part of the cell, a property that is not necessarily true for small molecules. Most strategies modify natural plant photoreceptors to create light sensitive proteins. One popular choice is the light oxygen voltage domain from phototropin (LOV2), which consists of a core flavin mononucleotide-binding domain followed by a C-terminal Jα helix.
Computational Redesign of Metalloenzymes for Catalyzing New Reactions
The developers of the online game Foldit (70) crowd-sourced solutions for the challenge of de novo protein design (71). Online Foldit players were provided with a set of tools to generate, mutate, move, and score protein structures. Starting from a fully extended peptide chain, players were able to fold the chain into de novo structures and stabilize the structures by sequence optimization.
Protein structures can be broken up into three-dimensional local pieces called tertiary structural motifs (TERMs) (133) (Fig. 4C). Half of the structures in the PDB can be described by only about 600 TERMs (37), indicating that the sequence preferences of each TERM could be used to calculate the fitness of a sequence for a given local structure. A strong correlation (133) was observed between the TERM-derived scores and protein structure model accuracies from the Critical Assessment of Structure Prediction.
Drawing inspiration from these natural marvels, we seek to design equally useful molecules from scratch. Schematic illustration of a systematic, minimal approach to the design of the four-helix bundle protein. Protein design weaves together principles from biology, chemistry, and physics, allowing researchers to create novel molecules with remarkable precision and functionality. Antibodies are proteins that recognize and neutralize foreign substances like bacteria and viruses, protecting us from infections. On the other hand, enzymes, which are also proteins, speed up countless chemical reactions in our bodies, allowing us to digest food, synthesize DNA, and produce energy.
Constrained peptides
By continuing to design protein building blocks that are more sensitive to stimuli such as pH, light, ionic strength, and temperature, we will expand the functionality of designed materials in the future. We still do not have the theoretical or computational tools to design any protein structure or any protein-protein interaction interface on demand. Wild-type fluorescent proteins are often oligomers, a property that could interfere with the natural activity of a protein of interest. A common design strategy to prevent oligomerization is to introduce unfavorable electrostatic interactions to disrupt the subunit interfaces.
Short fragments with desired secondary structures are then extracted from the PDB and assembled into a three-dimensional protein model (Fig. 2A). Top7 was the first protein designed by this method and has a fold topology not observed in nature (33). We reasoned that RFdiffusion might be able to address this challenge by directly generating binding proteins in the context of the target. For many therapeutic applications, for example, blocking a protein–protein interaction, it is desirable to bind to a particular site on a target protein. To enable this, we fine-tuned RFdiffusion on protein complex structures, providing a feature as input indicating a subset of the residues on the target chain (called ‘interface hotspots’) to which the diffused chain binds (Fig. 6a and Extended Data Fig. 8a,b).
Parameters such as sheet curvatures, loop types, and secondary structure lengths were sampled during a hierarchical backbone assembly process. Thousands of stable designs with diverse pocket geometries were identified by a high-throughput yeast surface display experiment. We solved the structure of the highest affinity Influenza binder, HA_20, in complex with Iowa43 HA using cryo-EM (Extended Data Table 1). Raw electron micrographs revealed a well-folded HA glycoprotein with clearly discernible side, top and tilted view orientations suspended in a thin layer of vitreous ice (Extended Data Fig. 9a). The 2D class averages further show clear secondary structure elements corresponding to both Iowa43 HA (Extended Data Fig. 9b), as well as the HA_20 binder bound to the stem (Fig 6e). The 3D heterogenous refinement without symmetry revealed full occupancy of all three HA stem epitopes by the HA_20 binder.
By simulating various sequences and analyzing the resulting structures, researchers can design proteins with desired functions or properties. In addition to the new algorithms’ power, the tremendous amount of structural data captured by biologists has also allowed the protein design field to take off. The Protein Data Bank, a critical resource for protein designers, now contains more than 200,000 experimentally solved structures. The AlphaFold 2 algorithm is also proving to be a game changer here in terms of providing training material and guidance for design algorithms.

Next, the sequence from the target TM protein is “threaded” onto either one of the helices—in this example, the right helix with green side-chains (middle). Last, the sequence and side-chain configurations for the anti-TM peptide (represented by the left helix) is chosen via iteration over amino acids and rotamer re-packing (right). From Yin, H.; Slusky, J. S.; Berger, B. W.; Walters, R. S.; Vilaire, G; Litvinov, R.I.; Lear, J. D.; Caputo, G. A.; Bennett, J. S.; DeGrado, W. F. Science 2007, 315(5820), 1817–1822. Naturally occurring proteins with the same fold topology can have distinct functions because of fine-tuned differences in the precise geometries of structural elements (74, 75).
Machine learning for functional protein design - Nature.com
Machine learning for functional protein design.
Posted: Thu, 15 Feb 2024 08:00:00 GMT [source]
For example, many viral glycoproteins are trimeric and symmetry matched arrangements of inhibitory domains can be extremely potent43,44,45,46. Conversely, symmetric presentation of viral epitopes in an arrangement that mimics the virus could induce new classes of neutralizing antibodies47,48. To explore this general direction, we sought to design trimeric multivalent binders to the SARS-CoV-2 spike protein. In previous work, flexible linkage of a binder to the ACE2 binding site (on the spike protein receptor binding domain) to a trimerization domain yielded a high-affinity inhibitor that had potent and broadly neutralizing antiviral activity in animal models43. Ideally, however, symmetric fusions to binders would be rigid, so as to reduce the entropic cost of binding while maintaining the avidity benefits from multivalency.
Today, AlphaFold 2 is used routinely by many structural biologists, with over 200 million structures predicted. We took advantage of the increased size of these oligomers (compared to the smaller unconditional and fold-conditioned monomers described above) and collected negative stain electron microscopy (nsEM) data on a subset of these designs across different symmetry groups. For most, distinct particles were evident with shapes resembling the design models in both the raw micrographs and subsequent two-dimensional (2D) classifications (Fig. 3 and Extended Data Fig. 5f).








No comments:
Post a Comment